The goal of this project is to develop manifold learning techniques for analyzing evolutionary spaces through geometric structure recovery methods. The project aims to experiment with embedding strategies and implement a pipeline for processing, embedding and visualizing gene sequences. Starting with mathematical problem statements for embedding to Euclidean, spherical and hyperbolic spaces, and a code snippet for semidefinite embedding, the objective is to create working implementations of these algorithms. The main task is to transform pairwise distances from phylogenetic trees into meaningful geometric embeddings that can reveal the structure of evolutionary data, making it possible to visualize relationships between genes and identify evolutionary patterns.
Report
The project successfully implemented four distinct embedding techniques and created a functional pipeline for genomic sequence analysis.
Key Results:
Implemented embedding algorithms: Classical MDS achieving zero RMSE for initially Euclidean data, spherical embedding achieving zero RMSE for initially spherical data using cosine Gram matrix and pivoted Cholesky decomposition, hyperbolic embedding achieving zero RMSE for initially hyperbolic data via Lorentzian eigendecomposition with Poincaré ball projection, and Maximum Variance Unfolding (MVU)
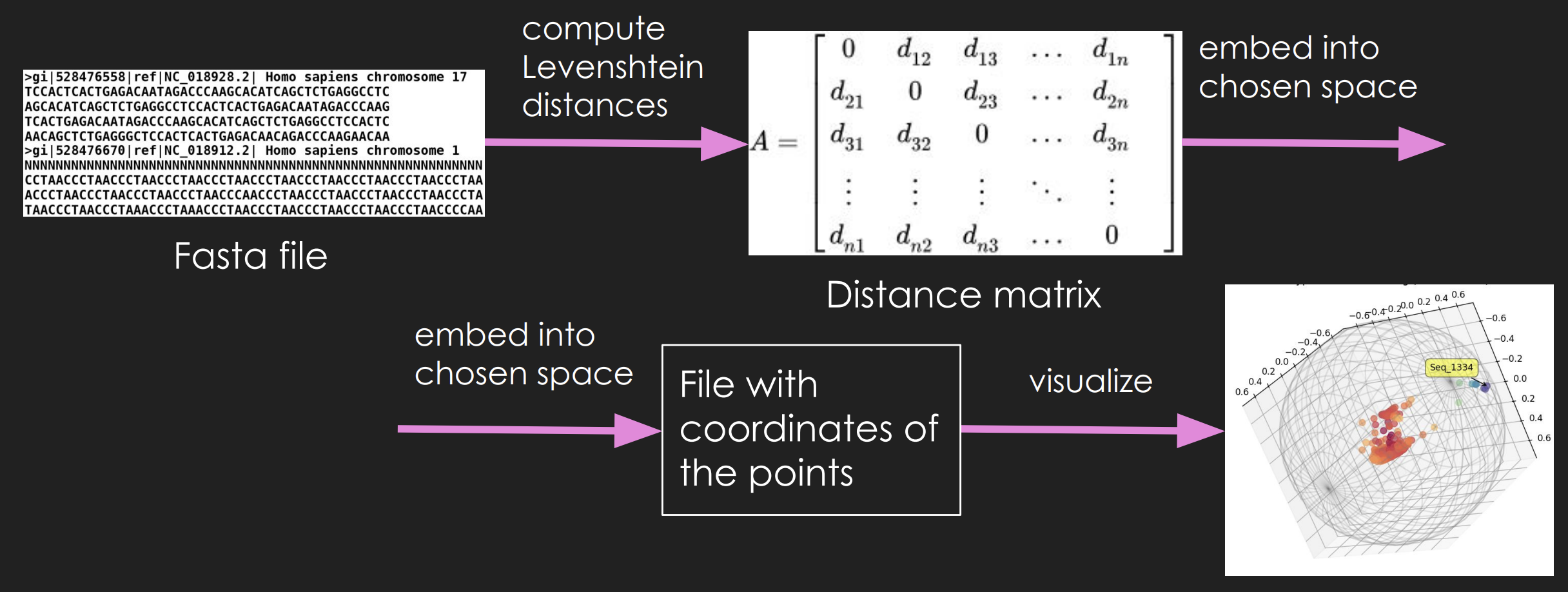
Developed genomic sequence pipeline: Created a system that processes FASTA files, computes pairwise Levenshtein or Hamming distances, embeds the distance matrix into chosen space (Euclidean, spherical, or hyperbolic), and visualizes results in 3D when dimension equals 3
Performance results: Benchmark tests on 100–500 points showed spherical embedding as fastest (0.0007–0.0065 seconds), while tests on genomic sequences (500–10,000 sequences) demonstrated Euclidean embedding processing times from 0.0663 to 93.1289 seconds with RMSE values increasing with dataset size
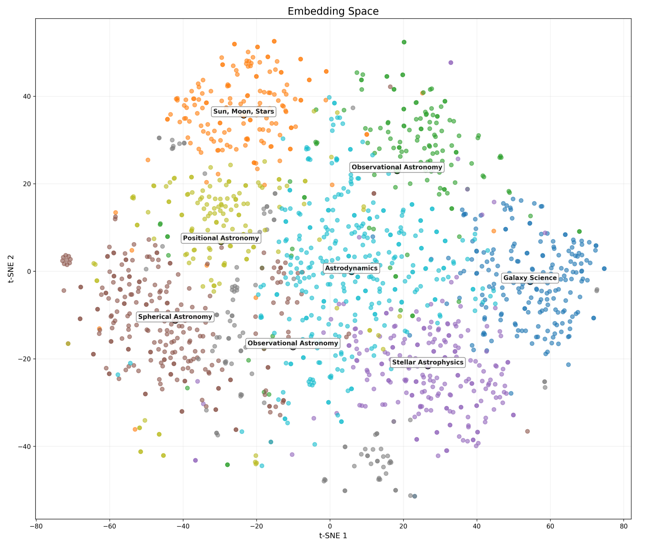
Practical applications demonstrated: The embeddings enable visualization where alike genes are placed closely to each other, reveal structural parameters of evolutionary space, and allow marking the probable location of the last common ancestor through identification of "evolutionary hollows"
Sources are available in GitHub repository.